【CMT&CHTV 文献精粹】
导语:本研究提出了一种基于领域泛化的深度学习算法Cancer-Finder,该算法能快速识别恶性细胞,对肿瘤异质性的研究和个性化治疗具有重要意义。
研究背景
肿瘤异质性是癌症治疗发展的重要障碍。单细胞RNA测序(scRNA-seq)技术通过在单细胞水平上揭示肿瘤的内部和间质异质性,推动了个性化治疗的发展。空间转录组学(ST)技术进一步捕捉了组织内部转录活性的空间背景,为癌症研究提供了新的视角。然而,现有的算法在恶性肿瘤细胞注释的准确性和泛化能力上存在不足,难以从泛癌数据中快速、一致地推断出恶性细胞。
2024年2月,Nature Communications杂志发表了题为“Domain Generalization Enables General Cancer Cell Annotation in Single-cell and Apatial Transcriptomics”的研究,旨在快速识别单细胞数据中的恶性细胞,并能够准确识别空间转录组学数据中的恶性斑点。

研究方法
该研究是一项基于深度学习的算法开发研究,旨在快速识别单细胞数据中的恶性细胞,并能够准确识别空间转录组学数据中的恶性斑点。研究团队收集了74个人体肿瘤微环境数据集作为训练集,并将其分为14个不同的类别。通过下采样确保了类别平衡,并使用风险外推的方法进行领域泛化训练,以提高模型在不同数据集上的泛化性能。
研究结果
Cancer-Finder在内部验证数据集上的平均准确率达到了95.16%,在外部验证数据集上的平均准确率为98.30%。此外,Cancer-Finder在空间转录组数据上也展现出了良好的预测能力,经过小规模训练集训练后,对训练过的组织的预测准确度在82.00-97.37%之间。研究还成功识别了一个由10个基因组成的基因签名,这些基因在肿瘤-正常组织界面显著共定位,并且与透明细胞肾癌患者的预后密切相关。
此外,Cancer-Finder在多个数据集上的表现均优于现有的其他工具,如SCEVAN、CopyKAT、CaSee和ikarus等。在对超过500 000个细胞的Cancer Single-cell Expression Map (CancerSCEM)数据库进行注释时,Cancer-Finder在一小时内就能快速预测,且与数据库提供的恶性细胞百分比高度相关。
Cancer-Finder算法的高准确率识别能力
该研究中,Cancer-Finder算法在单细胞转录组数据上展现了卓越的性能,其对恶性细胞的识别准确度达到了平均95.16%。这一结果远超过传统方法,如基于逻辑回归的Ikarus算法,以及基于拷贝数变异推断的CopyKAT算法。在5个透明细胞肾癌(ccRCC)的空间转录组样本测试中,Cancer-Finder同样表现出色,准确识别恶性斑点的能力,准确度在82.00%~97.37%之间(图1)。Cancer-Finder算法不仅在单一数据集上表现出色,它在跨癌种的数据集上同样能够保持高准确度的识别能力。在对5种不同透明细胞肾癌的空间转录组样本进行分析时,算法成功地识别出了恶性肿瘤区域,并且识别出的基因签名与肿瘤-正常组织界面显著相关。这些数据强有力地证明了Cancer-Finder算法在单细胞及空间转录组数据中的泛化能力。
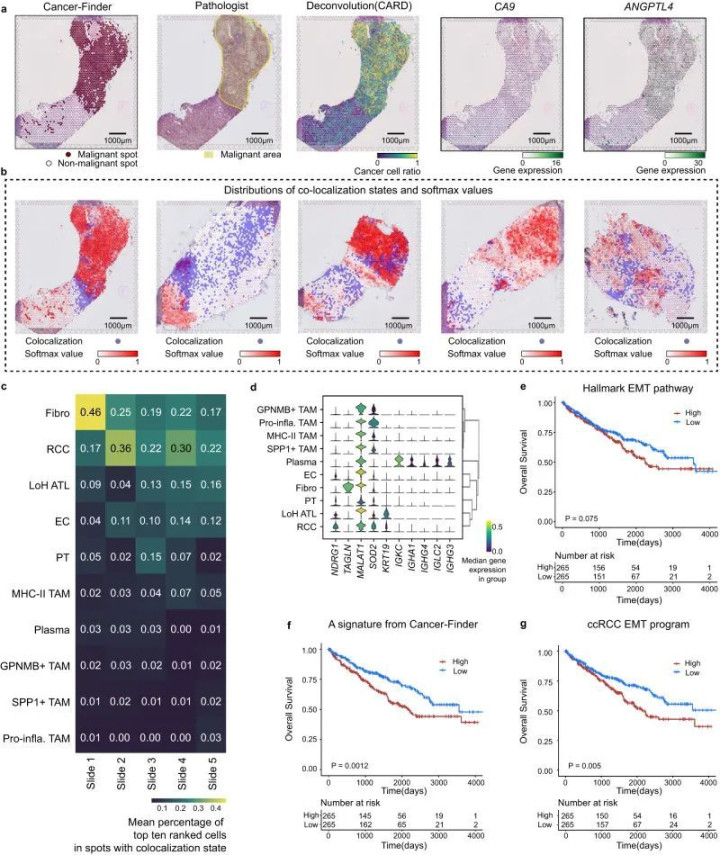
图1.Cancer-Finder在ccRCCST数据集中肿瘤间异质性分析中的应用
基因签名的发现及其临床意义
研究团队利用Cancer-Finder算法识别出的基因签名由10个基因组成,它们在肿瘤与正常组织界面显著共定位,并且与透明细胞肾癌患者的预后具有较强的相关性。通过单样本基因集富集分析(ssGSEA)计算这些基因的表达签名得分,发现这些基因在肿瘤-正常组织界面富集,且与患者的总体生存率显著相关。这一发现不仅为理解肿瘤微环境提供了新的视角,也可能为临床治疗提供了潜在的生物标志物。
Cancer-Finder算法在大规模数据集上的应用潜力
在对Cancer Single-cell Expression Map (CancerSCEM)数据库的超过500 000个细胞进行注释时,Cancer-Finder算法展现了其在大规模数据集上的应用潜力。它在一小时内就能完成快速预测,并且与数据库提供的恶性细胞百分比高度相关(Pearsons相关系数>0.85)。此外,Cancer-Finder在不同大小的数据集上的推断速度均优于其他方法,显示出其在处理大规模数据时的高效性。
算法的泛化能力与领域发展价值
Cancer-Finder算法的泛化能力不仅限于已训练的数据类型,还能够扩展到其他空间转录组学技术生成的数据集,如MERFISH、Slide-seq等。这一泛化能力使得Cancer-Finder算法在不同的技术平台上均能保持较高的准确度,从而为肿瘤微环境的多维度研究提供了强有力的工具。此外,Cancer-Finder算法的高效性和可扩展性,预示着其在肿瘤微环境研究以及临床应用中的巨大潜力,特别是在个性化医疗和精准治疗领域。
讨论与总结
该研究的独特价值在于Cancer-Finder算法的高效性和可扩展性。Cancer-Finder算法不仅能够处理单细胞RNA测序数据,还能够扩展到空间转录组学数据的注释,这对于理解肿瘤微环境的复杂性具有重要意义。此外,Cancer-Finder的推断速度快,内存消耗低,使其在大规模数据分析中具有潜在的应用前景。尽管在血液肿瘤数据上的表现有限,但该算法在实体瘤的研究中展现出了巨大的潜力,为未来肿瘤微环境的研究和临床应用提供了新的工具和视角。
参考文献
ZHONG Z, HOU J, YAO Z, et al. Domain generalization enables general cancer cell annotation in single-cell and spatial transcriptomics[J]. Nat Commun, 2024, 15(1):1929. DOI:10.1038/s41467-024-46413-6.
“医学论坛网”发布医学领域研究成果和解读,供专业人员科研参考,不作为诊疗标准,使用需根据具体情况评估。